On February 25, 2025, a six-month-old infant named KJ Muldoon became the first human being to receive a personalized CRISPR gene editing therapy. Born with severe carbamoyl phosphate synthetase 1 (CPS1) deficiency — a rare metabolic disorder that prevents the liver from converting toxic ammonia into harmless urea — KJ faced a stark prognosis. Without a functioning CPS1 enzyme, every protein meal, every fever, every common cold risked sending ammonia levels spiraling to brain-damaging, potentially fatal concentrations. He was too small for a liver transplant, the only conventional treatment. His doctors at Children’s Hospital of Philadelphia (CHOP) and Penn Medicine had six months to invent something that had never existed before: a gene editing therapy designed for a single patient’s unique mutation. They succeeded. One year later, KJ is walking, talking, and thriving. And three days before his anniversary, the FDA announced a regulatory framework that could make his story the first of thousands.
Days since first personalized CRISPR therapy
Incidence of CPS1 deficiency
Months from diagnosis to treatment
Americans with rare genetic diseases
I. The Disease: When Your Own Metabolism Becomes Poison
To understand what makes KJ’s treatment extraordinary, one must first understand what makes his disease so devastating.
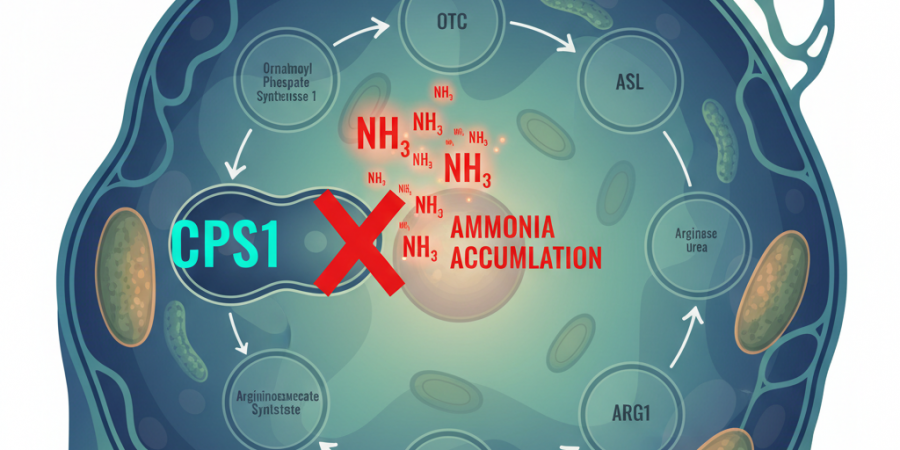
The urea cycle is one of the body’s most fundamental metabolic processes — a five-enzyme cascade that converts the ammonia produced by normal protein metabolism into urea, which is then harmlessly excreted through urine. It is, in essence, the body’s nitrogen waste disposal system. When it works, you never think about it. When it doesn’t, the consequences are catastrophic.
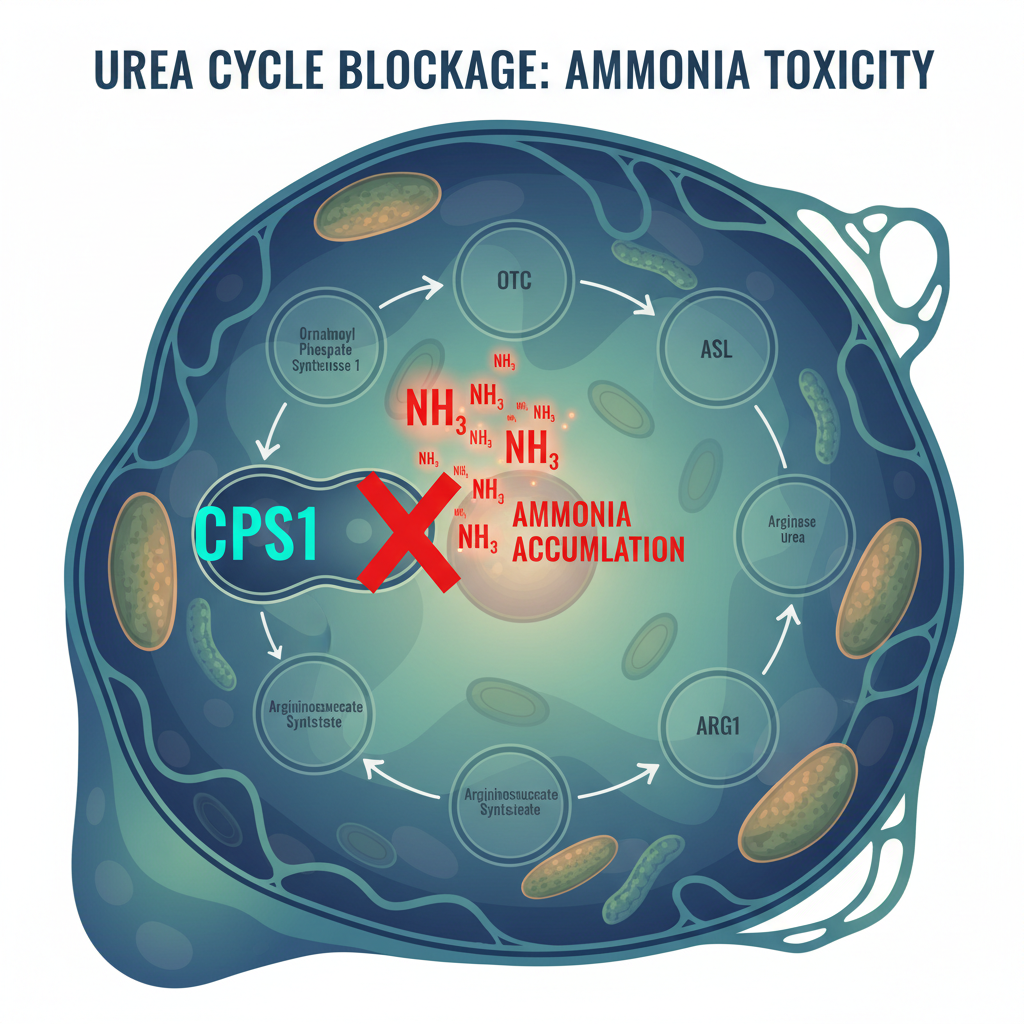
CPS1, or carbamoyl phosphate synthetase 1, catalyzes the first and rate-limiting step of this cycle. It is the gatekeeper. Located in the mitochondrial matrix of liver cells, CPS1 takes ammonia and bicarbonate and converts them to carbamoyl phosphate — the entry molecule for the entire urea cycle. Without functional CPS1, ammonia has nowhere to go. It accumulates in the blood, crosses the blood-brain barrier, and begins destroying neurons.
Severe CPS1 deficiency affects approximately 1 in 1.3 million births. Affected infants typically present within the first days of life with hyperammonemic crisis — lethargy, vomiting, seizures, and coma. Without immediate intervention, death follows within days. Those who survive the initial crisis face a life defined by extreme dietary protein restriction (often limiting natural protein intake to just 4–6 grams per day), nitrogen-scavenging medications that force alternative waste pathways, and the constant threat that any metabolic stress — a fever, an infection, even a growth spurt — could trigger another ammonia spike. The neurological damage from repeated hyperammonemic episodes is cumulative and irreversible.
KJ was diagnosed within days of birth and transferred to CHOP, where he spent the first months of his life in the NICU on a severely restricted diet, tethered to IV medications that kept his ammonia levels just below the danger threshold. His parents, Kyle and Nicole Muldoon, watched their son exist in a medical holding pattern — alive, but trapped.
“We would do anything for our kids, so with KJ, we wanted to figure out how we were going to support him and how we were going to get him to the point where he can do all the things a normal kid should be able to do,” Nicole Muldoon said. “We thought it was our responsibility to help our child.”
II. The Team: Twenty Years of Research Meets One Family’s Desperation
The therapy that would save KJ did not materialize overnight. It was the product of two decades of parallel research trajectories that converged at exactly the right moment.

Rebecca Ahrens-Nicklas, MD, PhD, directs CHOP’s Gene Therapy for Inherited Metabolic Disorders Frontier Program. Her career has been built on the premise that inborn errors of metabolism — diseases once considered untreatable at their root cause — could be corrected at the genetic level. Kiran Musunuru, MD, PhD, at Penn Medicine’s Perelman School of Medicine, came from the cardiovascular side. As a co-founder of Verve Therapeutics, he had pioneered the use of base editing — a refined form of CRISPR that changes individual DNA letters without cutting the double helix — to modify cholesterol genes in the liver. The two had begun collaborating in 2023 to study whether personalized base editing therapies could be created for individual patients with rare metabolic diseases.
When KJ arrived at CHOP, the pieces were already in place. Musunuru’s lab had extensive experience with lipid nanoparticle (LNP) delivery systems — the same technology that powered the COVID-19 mRNA vaccines — to ferry gene editing tools specifically to liver cells. Ahrens-Nicklas understood the metabolic disease landscape and the clinical pathway. What they needed was speed.
Within six months, the team identified KJ’s specific CPS1 mutation, designed a base editing guide RNA to correct it, manufactured the therapy, ran preclinical safety assessments, and obtained emergency FDA authorization. The editor was packaged into lipid nanoparticles — microscopic fat bubbles that are naturally taken up by liver cells — and administered intravenously. No viral vectors. No permanent DNA cuts. A single-letter correction delivered directly to the organ where it was needed.
“We took that same heart disease therapy and changed the GPS to turn the broken gene in his liver back on,” Musunuru explained. The elegance of the approach lay in its modularity: the delivery system and base editing machinery stayed the same. Only the targeting guide RNA — the molecular “GPS coordinates” — needed to change for each patient’s unique mutation.
III. The Results: What One Year of Data Shows
KJ received three infusions of the personalized therapy between February and April 2025. The results, published in the New England Journal of Medicine in May 2025, were cautiously described as “promising.” One year later, the clinical picture has become considerably clearer.

As of February 2026, KJ is walking, talking, and hitting developmental milestones. He tolerates significantly more dietary protein than before treatment. His dependence on nitrogen-scavenging medications has decreased. Most critically, he has weathered typical childhood illnesses — rhinovirus, minor infections — without the ammonia spikes that would have previously required emergency hospitalization.
“It’s amazing to see KJ hit these milestones,” Ahrens-Nicklas said at the one-year anniversary announcement. “While this treatment isn’t a cure, after three infusions from February through April 2025, KJ has tolerated it well with no serious side effects.”
The phrase “isn’t a cure” matters. KJ still requires monitoring, dietary management, and medical oversight. The base editing corrected his CPS1 mutation in a fraction of liver cells — enough to provide meaningful metabolic support, but not enough to fully restore normal urea cycle function. He will be followed for years to determine whether the correction is durable, whether additional doses might be needed, and whether long-term safety concerns emerge.
But the significance of KJ’s case extends far beyond one child’s medical trajectory. What his team demonstrated was not just a therapy — it was a method. A platform. A replicable process for creating individualized genetic medicines on a timeline measured in months rather than decades.
IV. The Platform: From N-of-1 to N-of-Many
The most transformative aspect of KJ’s treatment is not the treatment itself — it is the realization that the same process can be repeated, with modifications, for thousands of different mutations.

Consider the numbers. There are approximately 7,000 known rare diseases. Roughly 80% of them have a genetic basis. Collectively, they affect an estimated 30 million Americans and 300 million people worldwide. The vast majority have no approved treatment. The reason is brutally simple economics: pharmaceutical companies require large patient populations to justify the billion-dollar cost of traditional clinical trials. A disease that affects 50 people worldwide will never attract that investment.
KJ’s platform inverts this equation. Rather than developing an entirely new drug for each disease, the approach treats the base editor and delivery system as a reusable chassis. Only the guide RNA — a short sequence of nucleotides that directs the editor to the correct genomic location — changes between patients. Manufacturing a new guide RNA is relatively straightforward and inexpensive compared to developing a novel drug from scratch.
“We can do this over and over again with liver-centered diseases,” Musunuru stated. His team has already begun designing an umbrella clinical trial for urea cycle disorders — a single trial that would enroll patients with any of seven different genetic defects across seven different genes, all treatable by the same base editing platform with different guide RNAs.
The implications are staggering. A disease that affects 10 patients worldwide is no longer an economic impossibility. A mutation found in a single family is no longer beyond the reach of medicine. The marginal cost of treating the next patient drops dramatically once the platform infrastructure exists.
But there is a critical bottleneck: regulatory approval. And that bottleneck just cracked open.
V. The FDA’s Plausible Mechanism Pathway: A Regulatory Revolution
On February 23, 2026 — two days before KJ’s one-year anniversary — the FDA released draft guidance for what it calls the “Plausible Mechanism Pathway.” The timing was not coincidental. KJ’s case was explicitly cited as the inspiration.
The announcement was made at HHS headquarters in Washington, DC, hosted by HHS Secretary Robert F. Kennedy Jr. and FDA Commissioner Marty Makary, with Ahrens-Nicklas and Musunuru on stage alongside rare disease patient advocates. The core message was blunt: the traditional clinical trial framework is fundamentally incompatible with personalized medicine for ultra-rare diseases, and the FDA intends to change it.
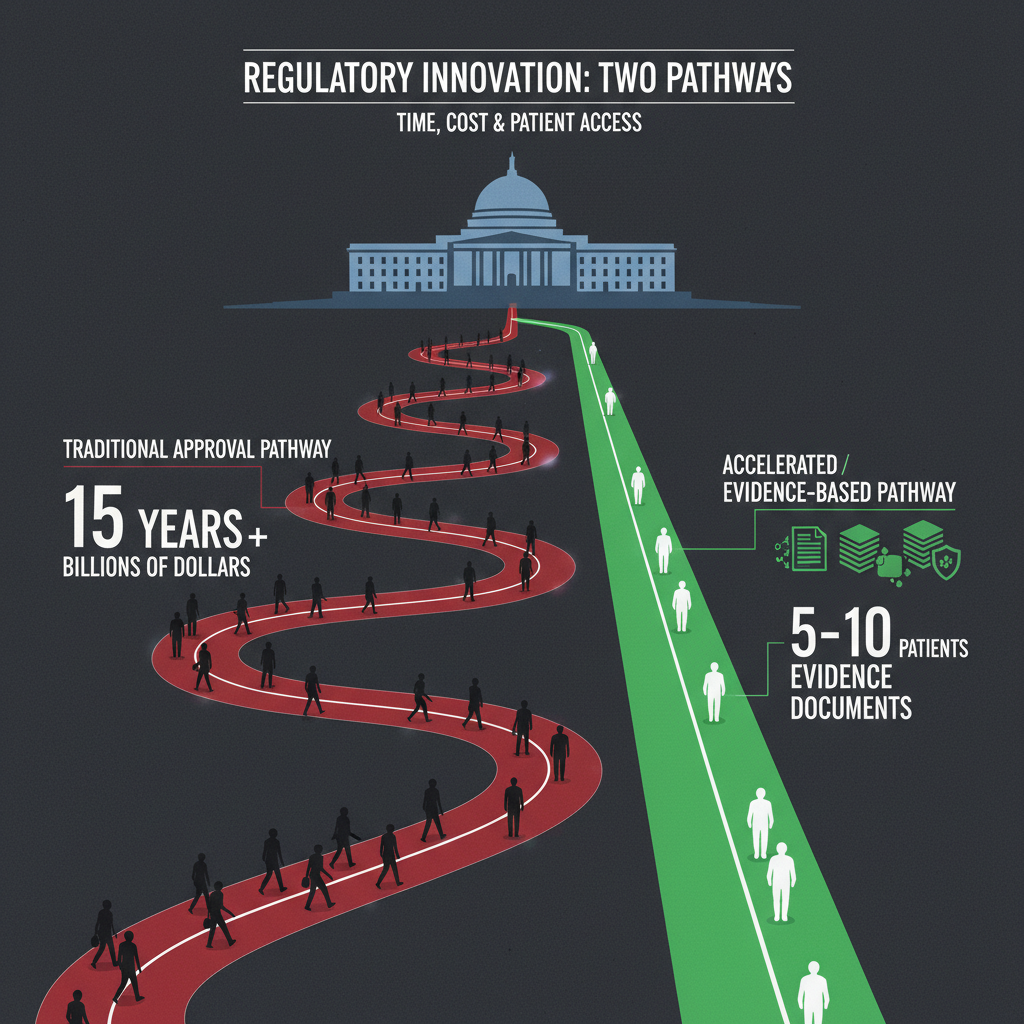
Under the traditional approval pathway, a new drug typically requires large randomized controlled trials with hundreds or thousands of patients to demonstrate statistical significance. For a disease that affects 50 people worldwide — scattered across different countries, ages, and medical systems — assembling such a trial is physically impossible. The requirement for large-scale statistical evidence, designed for common diseases, has become a barrier to treating rare ones.
The Plausible Mechanism Pathway offers an alternative. Instead of requiring large-scale trial data, the framework allows the FDA to evaluate therapies based on:
- Identification of the disease-causing genetic abnormality and evidence that correcting it should restore function (the “plausible mechanism”)
- Platform validation — evidence that the editing technology and delivery system work safely and effectively across multiple targets
- Small-cohort clinical data — positive results in as few as 5–10 patients, rather than hundreds, could support approval
- Unified drug classification — all variant-specific versions of a gene editor treated as a single drug, eliminating the need for separate trials for each mutation
“A disease with 100 distinct mutations in the same gene will no longer require 100 clinical trials,” Kennedy stated at the announcement. “Individualized medicine is no longer theoretical.”
Makary framed it as simple pragmatism: “Historically, rare diseases at the FDA have been an afterthought. Today, the majority of drugs we approve at FDA are for rare diseases. We need a framework that matches that reality.”
The draft guidance is now in a public comment period. If finalized in its current form, it would represent the most significant regulatory shift in rare disease medicine since the Orphan Drug Act of 1983.
VI. The Scale of the Challenge: 30 Million Americans Are Waiting
The raw numbers define the urgency. An estimated 30 million Americans — roughly 1 in 10 — live with a rare genetic disease. Fifteen million of them are children. For the vast majority, the treatment landscape consists of symptom management, supportive care, and waiting.

The term “rare disease” is itself somewhat misleading. While each individual condition may affect only a handful of patients, the aggregate population is enormous — larger than the combined populations of diabetes and heart disease. Yet rare disease research receives a fraction of the funding allocated to those common conditions. The NIH’s budget for rare disease research, while growing, remains disproportionately small relative to the patient population.
The result is a system that, as Judy Stecker — a rare disease parent advocate who spoke at the FDA announcement — described as driven by “desperate heroism.” Families raise millions through bake sales and golf tournaments. Parents become amateur geneticists, scouring PubMed for any researcher working on their child’s mutation. Individual families fund individual research programs for individual diseases — a model that, as Stecker noted, helps “one child, one mutation, one desperate family at a time.”
The Plausible Mechanism Pathway and the platform approach pioneered by KJ’s team offer the first realistic path to scaling beyond this model. If a single clinical trial can validate a platform across multiple mutations, and if regulatory approval can follow from small-cohort mechanistic evidence, then the cost and time barriers that have kept rare disease treatments out of reach begin to collapse.
“There are 30 million possible firsts in this country,” Stecker said at the HHS briefing. “Fifteen million are children, and one of them is mine.”
VII. The Manufacturing Puzzle: Can Bespoke Therapies Scale?
Even with a favorable regulatory framework, personalized gene therapies face a fundamental manufacturing challenge: each patient requires a unique guide RNA, custom quality control, and individualized production. The traditional pharmaceutical supply chain — built for producing millions of identical pills — is poorly suited to producing thousands of unique therapies.

The current state of the art, as demonstrated by KJ’s case, required six months from mutation identification to treatment administration. For a critically ill infant, six months is both a triumph of speed and an eternity of risk. Scaling to hundreds or thousands of patients per year will require dramatic compression of this timeline.
Several approaches are being explored. Automated guide RNA design using computational biology and machine learning could reduce the design phase from weeks to days. Standardized LNP manufacturing platforms could enable rapid encapsulation of new guide RNAs without reinventing the delivery system for each patient. And the FDA’s Plausible Mechanism Pathway, by treating variant-specific editors as components of a single approved platform, could eliminate much of the regulatory redundancy that currently slows each new application.
CHOP currently supports more than 80 faculty advancing cell and gene therapy across more than 20 programs and 45 active pediatric clinical trials. The infrastructure exists. The question is whether it can be replicated at centers worldwide, and whether the supply chain for individualized genetic medicines can achieve the reliability and speed that patients need.
Musunuru’s vision is explicit: “We aim to responsibly develop and scale these approaches, so more children can lead healthy lives, and to invest in hands-on training so best practices reach all communities.”
VIII. The Data Problem: Genomic Information in a Fragmented World
There is a less visible but equally critical challenge facing personalized medicine at scale: the genomic data infrastructure. Every personalized CRISPR therapy begins with a patient’s genetic sequence. That sequence must be accurately determined, securely stored, compared against reference databases, and shared with the research team designing the therapy — often across institutional and national boundaries.
The average diagnostic odyssey for a rare disease patient — the time from first symptoms to correct diagnosis — is five to seven years. This delay is rarely because the science is inadequate. In most cases, the patient’s mutation exists in a database somewhere, or could be identified through whole-exome sequencing. The bottleneck is informational: data siloed in hospital systems that don’t interoperate, genomic databases with inconsistent formats and access policies, and the fundamental challenge of matching a patient’s unique mutation against the sum of human genetic knowledge.
For personalized gene therapy to scale, this information architecture must evolve. Patients need the ability to maintain control over their genomic data while making it available for diagnostic matching and therapeutic design. Researchers need access to aggregated, anonymized datasets to validate editing strategies across mutation types. And the entire system must operate with the security and privacy guarantees that genomic information demands — this is, after all, the most personal data a human being possesses.
Decentralized data architectures — systems where individuals maintain sovereignty over their own information while enabling permissioned sharing — offer a compelling model. Platforms built on decentralized storage and cryptographically secured data exchange could allow a patient’s genomic sequence to be matched against global databases without ever leaving the patient’s control. GRIDNET OS, for instance, provides decentralized storage and crypto-incentivized data exchange infrastructure that could support exactly this kind of patient-controlled genomic data sharing, along with decentralized applications that give doctors and patients direct, secure interfaces for managing treatment data.
The genomic data problem is solvable. But solving it requires infrastructure designed for individual sovereignty rather than institutional convenience — a principle that the rare disease community, built on individual families fighting individual battles, understands intimately.
IX. Beyond the Liver: What Comes Next
KJ’s therapy targeted the liver — the organ where CPS1 functions and where LNP delivery systems naturally accumulate. But rare genetic diseases affect every organ in the body: the brain, the heart, the kidneys, the muscles, the eyes. Extending personalized gene editing beyond liver-centered diseases is the field’s next frontier, and it is a formidable one.
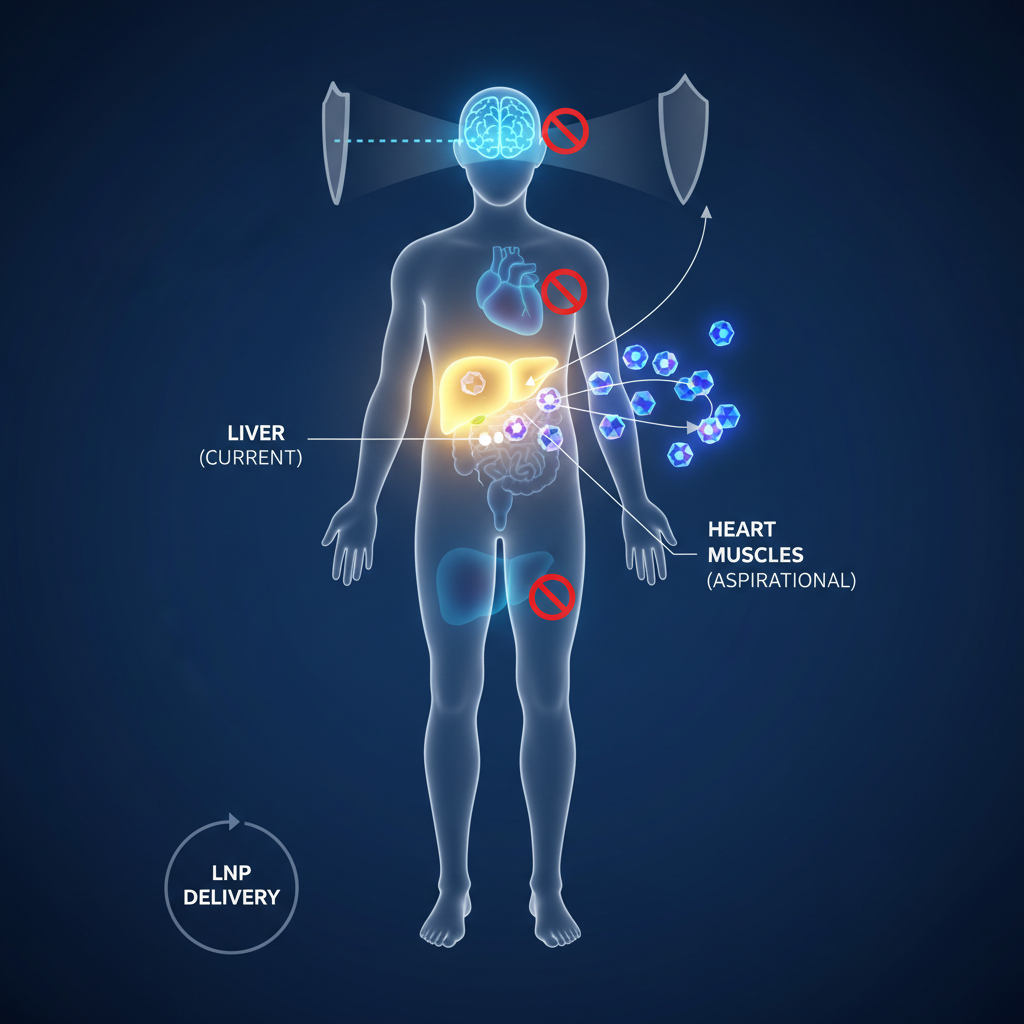
The blood-brain barrier remains the most formidable obstacle for neurological rare diseases — conditions like Batten disease, the disorder that killed Mila Makovec, the young girl whose 2018 treatment with an antisense oligonucleotide called Milasen became one of the earliest bespoke genetic therapies. Current LNP formulations do not efficiently cross the blood-brain barrier, meaning that diseases affecting the central nervous system require fundamentally different delivery approaches: intrathecal injection, engineered viral vectors with brain tropism, or novel nanoparticle formulations designed for CNS penetration.
Cardiac tissue presents different challenges — the heart is relatively accessible via the bloodstream, but cardiomyocytes are notoriously difficult to edit efficiently in vivo. Skeletal muscle, which constitutes 40% of body mass, requires delivery systems that can reach a vast tissue volume uniformly.
Each organ system demands its own delivery solution, its own safety profile, and its own clinical validation pathway. The platform concept — reusable editor, swappable guide RNA — applies to the editing machinery itself, but the delivery side of the equation may need to be organ-specific.
The research community is acutely aware of these challenges. Fyodor Urnov of the Innovative Genomics Institute and Jennifer Doudna’s co-founded Aurora Therapeutics are specifically targeting rare genetic disorders with next-generation delivery systems. David Liu, the inventor of base editing at the Broad Institute, has proposed treating 1,000 patients with rare genetic diseases by 2030 — a target that will require solutions for multiple organ systems.
X. The Moral Imperative: Who Gets Access?
The most uncomfortable question surrounding personalized gene therapy is not whether it works — KJ’s case demonstrates that it can — but who will be able to afford it.
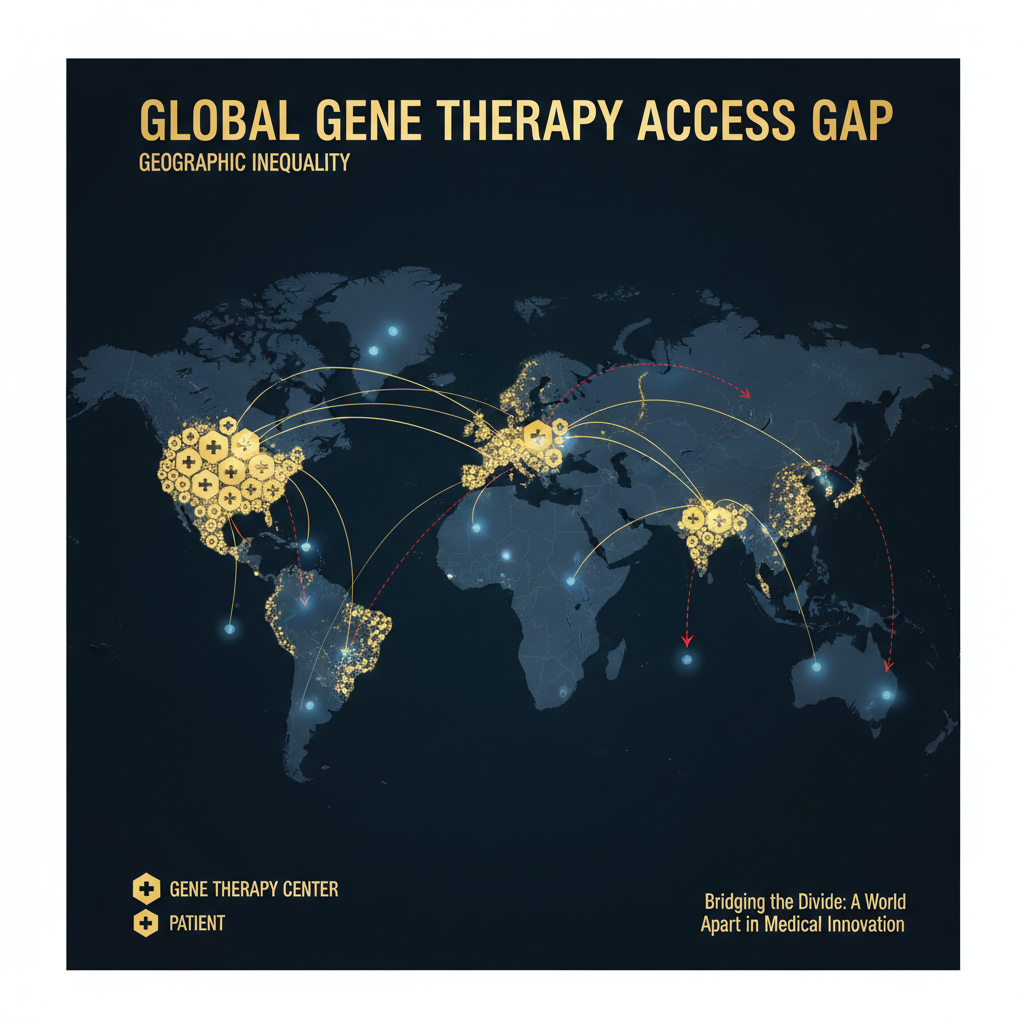
Casgevy and Lyfgenia, the first FDA-approved CRISPR therapies (for sickle cell disease), carry price tags of $2.2 million and $3.1 million per treatment respectively. These are one-size-fits-all therapies manufactured at scale for a relatively common genetic disease. A bespoke therapy designed for a single patient’s unique mutation, with custom manufacturing and individualized quality control, could easily exceed these costs — at least under current production methods.
The Plausible Mechanism Pathway addresses the regulatory barrier but does not solve the economic one. If each personalized therapy requires $500,000 to $2 million in manufacturing and quality control costs, the global population of rare disease patients — disproportionately located in countries without advanced healthcare infrastructure — faces a familiar injustice: the technology exists, but only for those who can pay.
Several forces could compress costs over time. Automated manufacturing, standardized platforms, and economies of learning (each new guide RNA design benefits from all previous ones) should drive marginal costs down as the field matures. Academic medical centers like CHOP, operating outside the profit-maximization logic of commercial pharma, may establish models where costs are absorbed through research funding and philanthropic support. And new models of decentralized research funding — where patient communities collectively invest in the development of therapies for their specific conditions — could distribute both the costs and the benefits more equitably.
But these are future possibilities. Today, the gap between what is medically possible and what is economically accessible remains the defining tension of the personalized medicine revolution.
“Every child deserves the same kind of treatment,” Kennedy said at the HHS briefing. The question is whether the systems we build — regulatory, economic, and technological — will make that statement a reality or leave it as rhetoric.
XI. One Year Later
On February 25, 2026, KJ Muldoon turned approximately 18 months old. He is walking. He is talking. He is eating more protein than his doctors ever thought possible. He has survived childhood illnesses that would have hospitalized him before treatment. His parents describe watching him grow as “nothing short of a miracle.”
Earlier this month, the Muldoon family traveled to Washington, DC, where they met with lawmakers alongside KJ’s doctors. CHOP hosted educational briefings with the Rare Disease Congressional Caucus and the Personalized Medicine Caucus. Kyle Muldoon told legislators: “We want to put a human face on rare diseases.”
The science that saved KJ was twenty years in the making. The regulatory framework that could make his treatment a template is three days old. The manufacturing infrastructure that could scale it is still being built. The economic models that could make it accessible are still being debated.
But KJ is walking. And in the world of rare disease medicine, where hope has been in desperately short supply for decades, that simple fact changes everything.
“We’re just beginning to unlock gene editing’s potential in pediatrics and beyond,” Musunuru said. “This will allow doctors to get treatments for many disorders.”
The promise of gene therapy, he added, “is coming to fruition, and it’s going to utterly transform the way we approach medicine.”
Thirty million Americans are waiting to find out if he’s right.
References & Sources
- Musunuru, K. et al. (2025). “Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease.” New England Journal of Medicine. DOI: 10.1056/NEJMoa2504747
- Children’s Hospital of Philadelphia (2026). “Children’s Hospital of Philadelphia Marks One-Year Anniversary of World’s First Personalized CRISPR Gene Therapy for Child with Rare Genetic Disease.” Press Release, February 25, 2026. chop.edu
- FDA (2026). “Considerations for the Use of the Plausible Mechanism Framework to Develop Individualized Therapies that Target Specific Genetic Conditions with Known Biological Cause.” Draft Guidance, February 23, 2026. fda.gov
- Makary, M. & Prasad, V. (2025). “A Plausible Mechanism Pathway for Bespoke Gene Therapies.” New England Journal of Medicine. DOI: 10.1056/NEJMsb2512695
- Children’s Hospital of Philadelphia (2025). “World’s First Patient Treated with Personalized CRISPR Gene Editing Therapy.” chop.edu
- STAT News (2026). “FDA unveils rules for bespoke gene therapies, predicting flood of rare disease applications.” February 23, 2026. statnews.com
- Inside Precision Medicine (2026). “FDA Issues Plausible Mechanism Pathway Draft Guidance to Spur Innovation for Individualized Therapies.” February 25, 2026. insideprecisionmedicine.com
- Congressional Research Service. “Cross-Border Data Sharing Under the CLOUD Act.” congress.gov
- Elabd, C. et al. (2014). “Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration.” Nature Communications, 5, 4082. DOI: 10.1038/ncomms5082
- National Organization for Rare Disorders (NORD). “Rare Disease Facts.” rarediseases.org
- American Society of Gene & Cell Therapy (2025). ASGCT Annual Meeting, New Orleans, May 2025.
- CBS Philadelphia (2026). “After a baby became the world’s first CRISPR gene editing therapy patient, CHOP wants to expand treatment to others.” February 26, 2026. cbsnews.com